CASE REPORT | https://doi.org/10.5005/jp-journals-10002-1264 |
Clinical Challenges in Nonfunctional Pheochromocytomas
1–3Department of Surgery, Division of Endocrine Surgery, National University Health System, Singapore
4Department of Pathology, Yong Loo Lin School of Medicine, National University of Singapore and National University Health System, Singapore
Corresponding Author: Rajeev Parameswaran, Department of Pathology, Yong Loo Lin School of Medicine, National University of Singapore and National University Health System, Singapore, Phone: +65 67724229, e-mail: rajeev_parameswaran@nuhs.edu.sg
How to cite this article Lui S-A, Oh HB, Tan KB, et al. Clinical Challenges in Nonfunctional Pheochromocytomas. World J Endoc Surg 2019;11(3):86–90.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Pheochromocytomas (PCCs) are rare endocrine tumors that arise from neural crest cells of the adrenal medulla. They commonly secrete catecholamines and other biological peptides that account for the symptoms namely, hypertension, palpitations, and episodic headaches associated with the condition. However, the symptoms and clinical presentations are highly variable due to variations in catecholamine biosynthesis and secretion because of differences in gene expression. A small proportion of tumors hardly synthesize or release any catecholamines and may have no symptoms and are termed as nonfunctional or subclinical PCCs. The nonfunctional tumors are commonly picked up as incidentalomas and biochemical work-up is usually negative. Undiagnosed subclinical normotensive PCCs could lead to catastrophic consequences during surgery and subsequently.
Keywords: Catecholamines, Incidentalomas, Pheochromocytoma.
INTRODUCTION
Pheochromocytomas (PCCs) (Greek phaios “dark”, chroma “color”) or adrenal paraganglioma (PG) is a neuroendocrine tumor arising from the chromaffin cells of the adrenal medulla.1 It is a sympathetic tumor with diverse manifestations due to variations in the catecholamine secretion and their metabolites, unlike the head and neck PGs, which are parasympathetic in origin and do not usually produce catecholamines. While most adrenal PCCs present with the symptoms of hypertension, palpitations, and paroxysmal headaches, about 14–55% of incidental lesions are asymptomatic.2,3 Pheochromocytomas are considered as “the great mimic” due to their diverse presentations, which can pose a challenge to clinicians. Here, we review two cases of PCCs, which presented as incidentalomas, completely asymptomatic, and histology revealed the presence of large PCCs.
CASE DESCRIPTION
Case 1
A 45-year-old man was admitted following a motorcycle accident with multiple lacerations and fractures of his right upper arm. During his admission, his blood pressure (BP) was completely normal, and he had no symptoms, apart from those related to his trauma. A whole-body computerized tomography scan showed a fracture along with a 5.6 cm right adrenal incidentaloma. The CT showed the lesion to be heterogeneous with areas of hypo-densities in keeping with necrosis (Fig. 1A). Biochemical workup in the form of urinary metanephrines and PCCs screen was completely normal. He underwent laparoscopic adrenalectomy with intraoperative BP fluctuations that were controlled with antihypertensive agents and postoperative recovery was uneventful. Histology showed the presence of PCCs with local capsular invasion and genetic screen revealed no mutations for an inherited disease.
Case 2
A 55-year old woman presented with symptoms of right upper quadrant pain and dyspepsia and backache. Ultrasound scan of the liver showed no obvious abnormalities apart from a mass measuring about 7 cm above her kidney and this was confirmed with a CT scan (Fig. 1B). Biochemical workup in the form of urinary metanephrines and PCCs screen was completely normal. She underwent open adrenalectomy without any complications during the perioperative and postoperative period; histology showed the presence of PCCs with a pheochromocytoma of the adrenal gland scaled score (PASS) of 4 (Figs 2A and B). A genetic screen showed no mutations for the inherited patterns of disease (Table 1).
DISCUSSION
Pheochromocytomas are catecholamine secreting neuroendocrine tumors arising from chromaffin cells of the adrenal medulla, with an incidence of about 0.1% of the general population and 0.1–0.2% of patients presenting with hypertension. Not until recently 90% of PCCs was believed to occur sporadically but with advances in genetic studies have shown that up to 25% of patients have an inherited pattern with germline mutations.4–6 Inherited PCCs are associated with conditions such as multiple endocrine neoplasia type II (MEN-2A or MEN-2B), von Recklinghausen’s neurofibromatosis type I (NF-1), von Hippel–Lindau (VHL) syndrome, and familial PG due to germline mutations of genes encoding succinate dehydrogenase (SDH) subunits B, C, and D.
Pheochromocytomas have been considered as a “great mimic” because of its protean manifestations (Table 2), from symptoms of progressive catecholamine excess leading to hypertension, which may be sustained and episodic, hypertensive crisis, cardiovascular manifestations, such as acute myocardial infarction and stroke. The symptoms associated with PCC is shown in Table 3. The most consistent sign in PCC is hypertension,7,8 as a result of activation of α- and β-adrenergic receptors by catecholamine excess. Besides catecholamines, many other vasoactive substances such as a calcitonin-gene-related peptide, vasoactive intestinal peptide, atrial natriuretic peptide, and neuropeptide Y (NPY) may be released, which may influence the response of the vasculature9,10 and this explains the fact that some patients are completely asymptomatic in the presence of catecholamine secreting tumor.7

Figs 1A and B: CT scan (coronal and sagittal) images of two patients showing a heterogeneous right adrenal mass
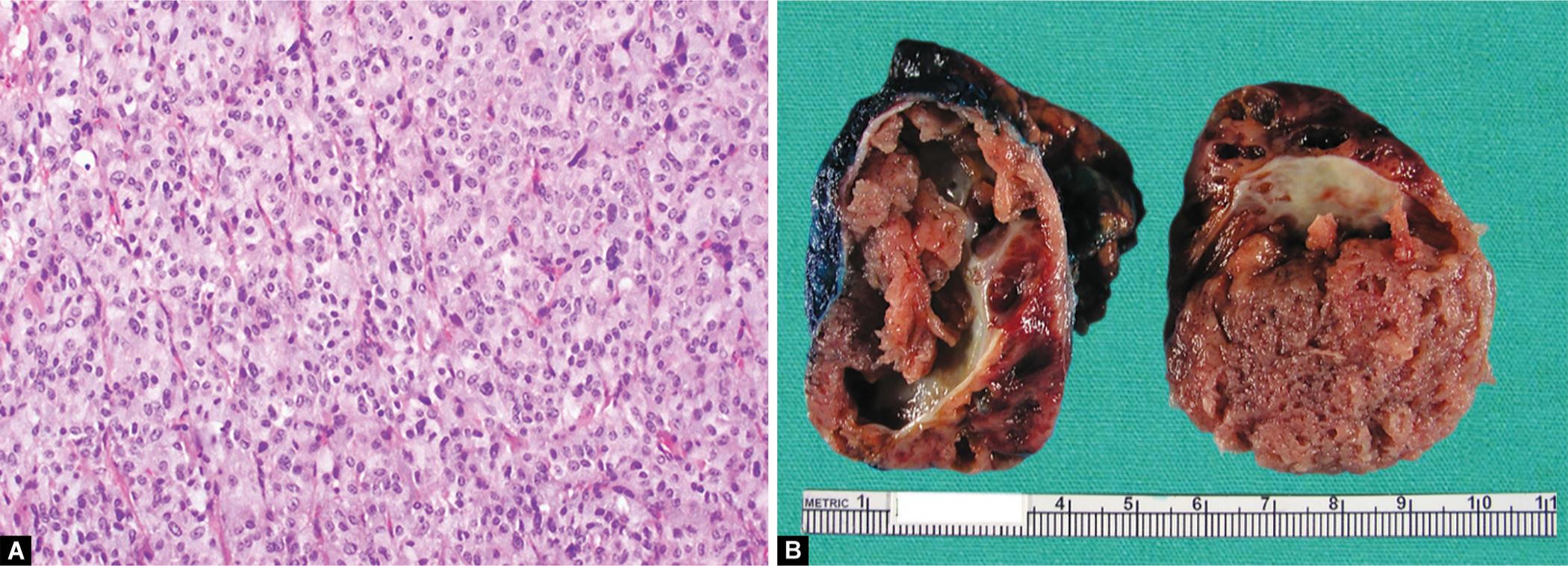
Figs 2A and B: (A) Resected adrenal gland is almost completely replaced by a well-circumscribed solid-cystic tumor, measuring 6 cm in maximum dimension; (B) Photomicrograph shows nests and trabeculae of tumor cells with ample, finely-granular cytoplasm, with intervening fine vasculature (hematoxylin and eosin, original magnification ×200)
Asymptomatic PCCs has been referred to as nonfunctional, silent or subclinical, and nonfunctioning or non-secreting.11–14 Absence of hypertension has been reported in 14–55% of incidental PCCs,2,15–17 especially those with adrenal incidentalomas and dopamine secreting tumors. Similarly, in patients with MEN2 syndrome, symptoms are only present in about 50% of patients and about 30% have hypertension.18 Recent study has shown that PCCs that are normotensive show a downregulation of five genes phenylethanolamine-N-methyltransferase (PNMT), secretogranin II, vesicular monoamine transporter type I, norepinephrine transporter, and NPY involved in key processes of catecholamine metabolism in comparison to those with hypertensive PCCs. This leads to small amounts of catecholamines that are secreted and thereby present with minimal clinical symptoms.17,19 Patients with nonfunctioning PCCs have a lower prevalence of diabetes suggesting a lower level of catecholamine synthesis compared with those with functioning tumors.20
| Age | Gender | Clinical presentation | Catecholamines 24-hour collection (nmol/day) | Values (nmol/day) | Size on CT (cm) | Histology |
|---|---|---|---|---|---|---|
| 45 | Male | Incidentaloma | Adrenaline (3–109) | 16 | 5.6 | PCC; sporadic; PASS 3 |
| Noradrenaline (89–473) | 296 | |||||
| Metanephrine (325–1,530) | 971 | |||||
| Normetanephrine (885–2,880) | 1,870 | |||||
| 44 | Female | Incidentaloma | Adrenaline (3–109) | 16 | 7 | PCC; sporadic |
| Noradrenaline (89–473) | 124 | PASS 4 | ||||
| Metanephrine (325–1,530) | 625 | |||||
| Normetanephrine (885–2,880) | 1,047 |
| Hyperthyroidism |
| Hypoglycemia |
| Panic disorder |
| Migraine |
| Carcinoid syndrome |
| Ischemic heart disease |
| Heart failure |
| Stroke |
| Arrhythmias |
| Epilepsy |
| Migraine |
| Symptoms | Incidence (%) | Signs | Incidence (%) |
|---|---|---|---|
| Headaches | 76–100 | Hypertension | 76–100 |
| Palpitations | 51–75 | Reflex bradycardia and tachycardia | 51–75 |
| Sweating | 51–75 | Hypotension | 51–75 |
| Anxiety | 26–50 | Weight loss | 26–50 |
| Abdominal/chest pain | 26–50 | Pallor | 26–50 |
| Weakness | 26–50 | Hyperglycemia | 26–50 |
| Tremulousness | 26–50 | Tremor | 26–50 |
Other factors that are associated with nonfunctional tumors besides the amount of catecholamines secreted are the nature of circulating catecholamines and the size of tumors. There are two types of phenotypes in PCCs—noradrenergic and adrenergic,21 which depends on the activity of PNMT, the enzyme that converts noradrenaline to adrenaline. Tumors that lack PNMT activity do not produce adrenaline and exhibit a noradrenergic phenotype.22 Both the phenotypes are found in both sporadic and tumors associated with germline mutations of identified 10 susceptibility genes namely: VHL,23RET,24NF1,25SDHA,26SDHB,27SDHC,28SDHD,29SDHAF2,30TMEM127,31 and MAX.32 The adrenergic phenotype is seen in tumors associated with mutations of RET, NF1, and TMEM127,21,33 whereas noradrenergic phenotype is seen VHL mutations.21
The size of the PCC also correlates with the amount of catecholamines and metanephrines, especially in patients who present with hypertension.34 This correlation is not seen in patients with asymptomatic PCCs. Similarly, large tumors with extensive hemorrhage and necrosis or in patients with SDHB mutations are also associated with mild symptoms or may be completely asymptomatic.35 A recent study has shown subclinical PCCs have reduced output of metanephrines because of impaired synthesis, storage, and release.17 The molecular signatures of subclinical PCCs are completely different from those of clinical PCCs due to differences in neuroendocrine differentiation and chromaffin cell phenotype.36,37
The question of the preoperative α blockade in patients with subclinical PCCs is debatable. North American Neuroendocrine Tumour Society consensus and Endocrine Society guidelines recommend that all patients with PCCs even with nonfunctional tumors should be given adequate α blockade prior to surgery.38,39 A study on 21 patients with nonfunctional PCCs who received preoperative α blocker showed no benefit in maintaining intraoperative hemodynamic instability and increased the use of colloids and vasoactive drugs.40 The potential for serious cardiovascular complications and mortality has been reported however, in patients with subclinical disease.41–45 Patients can sustain severe hypertensive crisis during surgery due to tumor manipulation, and significant postoperative hypotension following surgery.42 The operative duration appeared to be increased in functional PCCs compared with the nonfunctional PCCs, though the exact reason for this is not known.43 The risk of recurrence and malignancy in subclinical PCCs in comparison to clinical PCCs is not very well studied and further understanding of the unique molecular features will help understand the biology of subclinical tumors.
To summarize, subclinical PCCs can pose a significant challenge to clinicians and commonly present as adrenal incidentalomas. Pheochromocytomas with normotension have a distinct clinical, biological, and molecular characteristic distinct to PCCs that present with hypertension. There is also a difference in the concentration of urinary catecholamines between the two types of PCCs. The use of preoperative α blockade in nonfunctioning is debatable but may help manage hemodynamic instability. The long-term outcome of subclinical PCCs needs further evaluation.
REFERENCES
1. Lenders JW, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet. 2005;366(9486):665–675. DOI: 10.1016/S0140-6736(05)67139-5.
2. Kopetschke R, Slisko M, Kilisli A, et al. Frequent incidental discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma. Eur J Endocrinol 2009;161(2):355–361. DOI: 10.1530/EJE-09-0384.
3. Noshiro T, Shimizu K, Watanabe T, et al. Changes in clinical features and long-term prognosis in patients with pheochromocytoma. Am J Hypertens 2000;13(1 Pt 1):35–43. DOI: 10.1016/S0895-7061(99)00139-9.
4. Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005;23(34):8812–8818. DOI: 10.1200/JCO.2005.03.1484.
5. Neumann HP, Cybulla M, Shibata H, et al. New genetic causes of pheochromocytoma: current concepts and the clinical relevance. Keio J Med 2005;54(1):15–21. DOI: 10.2302/kjm.54.15.
6. Neumann HP, Berger DP, Sigmund G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med 1993;329(21):1531–1538. DOI: 10.1056/NEJM199311183292103.
7. Bravo EL. Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev 1994;15(3):356–368. DOI: 10.1210/edrv-15-3-356.
8. Bravo EL, Tagle R. Pheochromocytoma: state-of-the-art and future prospects. Endocr Rev 2003;24(4):539–553. DOI: 10.1210/er.2002-0013.
9. Besser GM, Thorner MO. Comprehensive clinical endocrinology,Edinburgh: Mosby; 2002.
10. Fonseca V, Bouloux PM. Phaeochromocytoma and paraganglioma. Baillieres Clin Endocrinol Metab 1993;7(2):509–544. DOI: 10.1016/S0950-351X(05)80186-7.
11. Leong CH, Wong KK, Saw D. Asymptomatic phaeochromocytoma of the bladder co-existing with carcinoma. Br J Urol 1976;48(2):123–126. DOI: 10.1111/j.1464-410X.1976.tb02996.x.
12. Krane NK. Clinically unsuspected pheochromocytomas. Experience at Henry Ford Hospital and a review of the literature. Arch Intern Med 1986;146(1):54–57. DOI: 10.1001/archinte.1986.00360130064008.
13. Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc 1981;56(6):354–360.
14. Simpson ET, Marszalek WW, Ramsaroop R. Nonfunctioning pheochromocytoma. Urology 1985;25(6):632–633. DOI: 10.1016/0090-4295(85)90299-7.
15. Mannelli M, Ianni L, Cilotti A, et al. Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol 1999;141(6):619–624. DOI: 10.1530/eje.0.1410619.
16. Mantero F, Terzolo M, Arnaldi G, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 2000;85(2):637–644. DOI: 10.1210/jc.85.2.637.
17. Haissaguerre M, Courel M, Caron P, et al. Normotensive incidentally discovered pheochromocytomas display specific biochemical, cellular, and molecular characteristics. J Clin Endocrinol Metab 2013;98(11):4346–4354. DOI: 10.1210/jc.2013-1844.
18. Eng C, Mulligan LM, Smith DP, et al. Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin Endocrinol 1995;43(1):123–127. DOI: 10.1111/j.1365-2265.1995.tb01903.x.
19. Zuber SM, Kantorovich V, Pacak K. Hypertension in pheochromocytoma: characteristics and treatment. Endocrinol Metab Clin North Am 2011;40(2):295–311. DOI: 10.1016/j.ecl.2011.02.002 ,vii.
20. Stenstrom G, Sjostrom L, Smith U. Diabetes mellitus in phaeochromocytoma. Fasting blood glucose levels before and after surgery in 60 patients with phaeochromocytoma. Acta Endocrinol (Copenh) 1984;106(4):511–515. DOI: 10.1530/acta.0.1060511.
21. Eisenhofer G, Walther MM, Huynh TT, et al. Pheochromocytomas in von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001;86(5):1999–2008. DOI: 10.1210/jcem.86.5.7496.
22. Eisenhofer G, Huynh TT, Pacak K, et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer 2004;11(4):897–911. DOI: 10.1677/erc.1.00838.
23. Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993;260(5112):1317–1320. DOI: 10.1126/science.8493574.
24. Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996;276(19):1575–1579. DOI: 10.1001/jama.1996.03540190047028.
25. White R, Viskochil D, O’Connell P. Identification and characterization of the gene for neurofibromatosis type 1. Curr Opin Neurobiol 1991;1(3):462–467. DOI: 10.1016/0959-4388(91)90070-N.
26. Burnichon N, Briere JJ, Libe R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet 2010;19(15):3011–3020. DOI: 10.1093/hmg/ddq206.
27. Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 2001;69(1):49–54. DOI: 10.1086/321282.
28. Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 2000;26(3):268–270. DOI: 10.1038/81551.
29. Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000;287(5454):848–851. DOI: 10.1126/science.287.5454.848.
30. Bayley JP, Kunst HP, Cascon A, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol 2010;11(4):366–372. DOI: 10.1016/S1470-2045(10)70007-3.
31. Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 2010;42(3):229–233. DOI: 10.1038/ng.533.
32. Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 2011;43(7):663–667. DOI: 10.1038/ng.861.
33. Yao L, Schiavi F, Cascon A, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA 2010;304(23):2611–2619. DOI: 10.1001/jama.2010.1830.
34. Eisenhofer G, Lenders JW, Goldstein DS, et al. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem 2005;51(4):735–744. DOI: 10.1373/clinchem.2004.045484.
35. Timmers HJ, Kozupa A, Eisenhofer G, et al. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 2007;92(3):779–786. DOI: 10.1210/jc.2006-2315.
36. Eisenhofer G, Pacak K, Huynh TT, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocr Relat Cancer 2011;18(1):97–111. DOI: 10.1677/ERC-10-0211.
37. Burnichon N, Vescovo L, Amar L, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet 2011;20(20):3974–3985. DOI: 10.1093/hmg/ddr324.
38. Chen H, Sippel RS, O’Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas 2010;39(6):775–783. DOI: 10.1097/MPA.0b013e3181ebb4f0.
39. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99(6):1915–1942. DOI: 10.1210/jc.2014-1498.
40. Shao Y, Chen R, Shen Z-J, et al. Preoperative alpha blockade for normotensive pheochromocytoma: is it necessary? J Hypertens 2011;29(12):2429–2432. DOI: 10.1097/HJH.0b013e32834d24d9.
41. Song G, Joe BN, Yeh BM, et al. Risk of catecholamine crisis in patients undergoing resection of unsuspected pheochromocytoma. Int Braz J Urol 2011;37(1):35–40; discussion-1. DOI: 10.1590/S1677-55382011000100005.
42. Shen SJ, Cheng HM, Chiu AW, et al. Perioperative hypertensive crisis in clinically silent pheochromocytomas: report of four cases. Chang Gung Med J 2005;28(1):44–50.
43. Lafont M, Fagour C, Haissaguerre M, et al. Per-operative hemodynamic instability in normotensive patients with incidentally discovered pheochromocytomas. J Clin Endocrinol Metab 2015;100(2):417–421. DOI: 10.1210/jc.2014-2998.
44. Yu R, Nissen NN, Chopra P, et al. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009;122(1):85–95. DOI: 10.1016/j.amjmed.2008.08.021.
45. Hariskov S, Schumann R. Intraoperative management of patients with incidental catecholamine producing tumors: A literature review and analysis. J Anaesthesiol Clin Pharmacol 2013;29(1):41–46. DOI: 10.4103/0970-9185.105793.
________________________
© The Author(s). 2019 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.