CASE REPORT |
https://doi.org/10.5005/jp-journals-10002-1422 |
Double Trouble: A Case of a Composite Pheochromocytoma
1Department of Critical Care, Chris Hani Baragwanath Hospital, Johannesburg, Gauteng, South Africa
2,3Division of Endocrinology, Department of Medicine, Chris Hani Baragwanath Hospital, Johannesburg, Gauteng, South Africa
4Division of Endocrine Surgery, Department of Surgery, Chris Hani Baragwanath Hospital, Johannesburg, Gauteng, South Africa
Corresponding Author: Imtiaz A Bahemia, Department of Critical Care, Chris Hani Baragwanath Hospital, Johannesburg, Gauteng, South Africa, Phone: +27795427967, e-mail: imt7@hotmail.com
ABSTRACT
Introduction: Pheochromocytomas are rare neuroendocrine tumors that arise from the chromaffin cells of sympathetic or parasympathetic tissue. Composite pheochromocytomas have additional histological components and account for only 3% of pheochromocytomas. We present a case of composite pheochromocytoma with chronic diarrhea, profound hypokalemia, and global weakness on presentation.
Case description: The patient reported an 18-month history of severe vomiting and watery diarrhea, with no associated headaches or sweating. She had an initial serum potassium of 1.1mmol/L (3.5–5.3), requiring a continuous intravenous potassium infusion, intubation, and ventilation for profound weakness. She remained normotensive throughout. A computed tomography (CT) scan revealed a left adrenal mass. She had markedly elevated urine metanephrine and normetanephrine levels [19,823 nmol/24 hr (152–913) and 17,933 nmol/24 hr (262–2129), respectively]. The patient underwent a left adrenalectomy and had complete resolution of her symptoms postoperatively. Histology revealed a composite tumor comprising both pheochromocytoma and ganglioneuroma.
Conclusion: This case highlights a rare presentation of pheochromocytoma, being an unusual cause of hypokalemia as well as interesting histopathology.
How to cite this article: Bahemia IA, Thomas TS, Mahyoodeen NG, et al. Double Trouble: A Case of a Composite Pheochromocytoma. World J Endoc Surg 2022;14(1):27-30.
Source of support: Nil
Conflict of interest: None
Keywords: Composite, Diarrhea, Ganglioneuroma, Hypokalemia, Pheochromocytoma
BACKGROUND
Pheochromocytomas are rare neuroendocrine tumors that arise from the chromaffin cells of sympathetic or parasympathetic tissue. The incidence is 0.6 per 100,000 person-years.1 The classic triad of symptoms are headaches, sweating, and palpitations. However, the clinical presentation is broad and atypical symptomatology and unusual histology may occur. Composite pheochromocytomas account for approximately 3% of pheochromocytomas. These tumors have additional histological components usually comprising neurogenic tumors such as neuroblastoma, ganglioneuroblastoma, ganglioneuroma, or other neuroendocrine neoplasms.2,3 They often present with atypical symptomatology if the coexisting tumor is hormonally active. Composite tumors share characteristics similar to conventional pheochromocytomas in that they can metastasize and be associated with hereditary syndromes. Literature has identified just over 70 cases to date and certain features seem more prominent: tumors appear to occur in older individuals and are generally of a larger size. The most common composite tumor is a ganglioneuroma.4 We present a case of a young female with a left adrenal composite pheochromocytoma who initially presented with chronic diarrhea, profound hypokalemia, and global weakness.
CASE DESCRIPTION
A 24-year-old female, with no known comorbidities, presented with an 18-month history of severe vomiting and watery diarrhea that was progressively increasing in frequency, despite periods of fasting. During this illness she had lost approximately 35 kg. Clinically she demonstrated profound global muscle weakness, an altered level of consciousness, and type II respiratory failure necessitating intubation and ventilation in the intensive care unit (ICU). Her initial serum potassium was 1.1 mmol/L (Table 1). She reported two prolonged admissions at other hospitals over the last year, with significant hypokalemia that would recur on discharge as diarrhea persisted. No cause had been elucidated at these facilities.
| Initial | Preoperative | Ref. range | |
|---|---|---|---|
| White blood cells (×109/L) | 20.36 | 15 | 3.9–12.6 |
| Creatinine (µmol/L) | 231 | 64 | 49–90 |
| Potassium (mmol/L) | 1.1 | 3.6 | 3.5–5.1 |
| Magnesium (mmol/L) | 0.561 | 1.28 | 0.63–1.05 |
| Chloride (mmol/L) | 85 | 107 | 98–107 |
| Urea (mmol/L) | 10.3 | 1 | 2.1–7.1 |
| C-reactive protein (mg/L) | 37 | 2 | <10 |
| Creatinine kinase (U/L) | 238 | 20–180 | |
| Normetanephrine (nmol/24 hr) | 17,933 | 562–2129 | |
| Metanephrine (nmol/24 hr) | 19,823 | 152–913 | |
| Chromogranin A (ng/mL) | 1397 | <101.9 | |
| Urine 5HIAA (µmol/24 hr) | 16 | 10.5–36.6 | |
| Aldosterone/direct renin ratio (pmol/mIU) | 7.8 | >75 is suggestive of hyperaldosteronism | |
| Stool microscopy and culture | Negative for Clostridium difficile, ova, cysts, and parasites | ||
In ICU, the patient required a continuous intravenous infusion of potassium to maintain normokalemia, despite correction of hypomagnesemia. She was extubated 48 hours after initial presentation. Primary aldosteronism was ruled out with an aldosterone/renin ratio below threshold in the face of a corrected potassium. Stool microscopy and culture, HIV enzyme-linked immunosorbent assay, urine 5-hydroxylindolacetic acid (5HIAA), gastroscopy, and colonoscopy were normal. In view of the persistent gastrointestinal symptoms, a CT abdomen was performed which demonstrated a large left adrenal tumor (12 cm × 10 cm) (Fig. 1). The 24-hour urine metanephrines were markedly elevated confirming the diagnosis of pheochromocytoma [metanephrines 19,823 nmol/24 hr (152–913) and normetanephrines 17,933 nmol/24 hr (562–2129)]. Of note, the patient remained normotensive throughout admission.

Fig. 1: Computed tomography scan of the abdomen demonstrating a 12 × 10 cm heterogeneous left adrenal mass (white arrow) with central necrosis and enhancement of 57 HU
The patient underwent an uncomplicated open left adrenalectomy. An adrenal tumor weighing 570 gm was excised (Fig. 2). She was preoperatively prepared with intravenous fluids, however, did not tolerate alpha blockade due to symptomatic hypotension. Histology revealed a composite tumor comprising both pheochromocytoma and ganglioneuroma cells, with negative staining for an SDHB mutation (Figs 3A and B and 4A to D). The pheochromocytoma of the adrenal gland scaled score was 2/21, signifying a low risk of malignancy.5 She had complete resolution of her symptoms after surgery and follow-up serum potassium levels remained in the normal range. A gallium dotatate positron emission tomography CT performed 6 weeks after surgery showed no evidence of residual tumor or metastases.
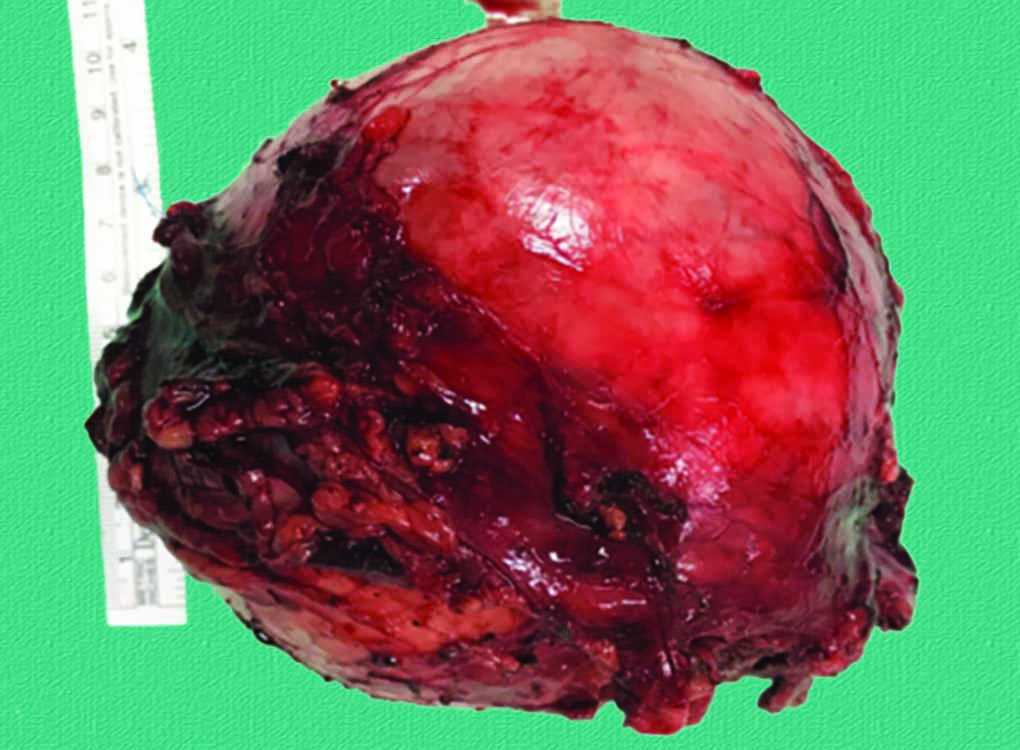
Fig. 2: Macroscopic adrenal tumor after resection weighing 570 gm, size of 11 × 9 × 6 cm with extensive necrosis and showing no evidence of capsular invasion
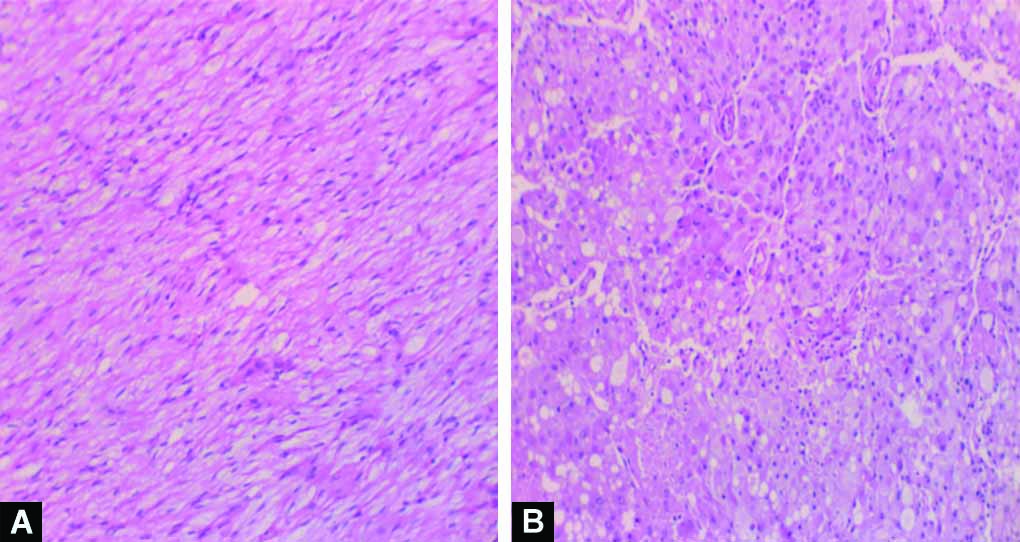
Figs 3A and B: Hematoxylin and eosin stain showing composite tumor with ganglioneuroma comprising ganglion and Schwann cells (Panel A) and pheochromocytoma comprising nests of polygonal cells separated by delicate vasculature (Panel B)

Figs 4A to D: Immunohistochemistry demonstrating both ganglioneuroma component (Panels A and C), neurofilament positive and only weakly positive for chromogranin A and pheochromocytoma components (Panels B and D), neurofilament negative and strongly positive for chromogranin A
DISCUSSION
We describe a young female who presented with chronic watery diarrhea and severe hypokalemia, and was found to have a composite pheochromocytoma. This case is atypical in that the patient presented without hypertension, but with severe hypokalemia which necessitated ICU care. Secondly, the histology demonstrated a composite tumor with both pheochromocytoma and ganglioneuroma components. Ganglioneuroma–pheochromocytomas are among the most common composite pheochromocytomas, accounting for about 65% of cases.6 Ganglioneuromas are often nonsecretory but a few cases associated with pheochromocytomas have been histologically confirmed to secrete vasoactive intestinal peptide (VIP) and can present with watery diarrhea hypokalemia achlorhydria (WDHA) syndrome, a syndrome most commonly observed with pancreatic tumors.7-9 Only 25 cases of pheochromocytoma with WDHA have been reported,10 with only 10 reported cases specifically noting a composite pheochromocytoma in conjunction with WDHA.11 Only one case has been reported in Africa.12 Composite pheochromocytomas have occasionally been found in association with an underlying genetic syndrome such as von Hippel Lindau and multiple endocrine neoplasia 2A. A systematic review on composite pheochromocytomas reported 28% of cases being associated with a genetic syndrome with only 2.2% of all composite pheochromocytoma presenting with WDHA.6
This patient presented with watery diarrhea with complete resolution of her symptoms after tumor removal. Management followed recent recommendations from guidelines: preoperative alpha blockade (although not tolerated due to hypotension), surgical removal with the least invasive techniques, and annual surveillance thereafter in the absence of multifocal or metastatic disease.1,13,14. The hypokalemia in this patient was most likely secondary to the profuse gastrointestinal losses and may have been further exacerbated by intracellular shifts of potassium in response to increased serum catecholamines. Her blood pressure was not elevated as she had severe volume depletion at presentation. However, even after fluid replacement she remained normotensive. We postulate that raised VIP levels may have contributed to vasodilation, thus counteracting the hypertensive effects of the elevated metanephrines. The patient was also unable to tolerate alpha blockade which potentially further supports the postulate of the vasodilatory effects of VIP.
The spectrum of the phenotypic presentation of pheochromocytoma is expanding. The wide variation of clinical symptomatology has been acknowledged in recent literature, with fewer patients presenting with the classic triad and a large proportion being asymptomatic (10–15%) or discovered incidentally on imaging (4–5%).1,15,16 Cases of normotensive pheochromocytoma were reported in 13.5–55% of incidental tumors,17 Haissaguerre et al. reported that normotensive pheochromocytoma may indeed represent a biochemical category of tumor different to hypertensive pheochromocytoma. In this case, functional imaging was not performed preoperatively as the bulky tumor would likely concentrate all of the radiotracer and not be accurately representative of multifocal or metastatic disease. Postoperative functional imaging excluded the latter conditions.
This case is not without limitations. Despite the clinical presentation suggesting VIP production, we were unable to test VIP levels as patient was on a proton pump inhibitor (PPI) which could falsely elevate results or perform immunohistochemical confirmatory staining due to a country-wide lack of the required reagent. In addition, we were unable to confirm the presence of achlorhydria due to administration of a PPI during the ICU stay.
This case highlights a rare histological sub-type of pheochromocytoma: the composite tumor. It also emphasizes that a high index of suspicion for the condition must be maintained in a patient with an unusual presentation, with severe hypokalemia being a recognized, albeit rare presentation of pheochromocytomas.
ETHICAL APPROVAL
This study was approved by the Human Research Ethics Committee (HREC) (Medical) of the University of the Witwatersrand (M210286).
GUARANTOR
Imtiaz A Bahemia.
ORCID
Imtiaz A Bahemia https://orcid.org/0000-0003-0884-1026
Teressa S Thomas https://orcid.org/0000-0003-1803-8392
Nasrin Goolam Mahyoodeen https://orcid.org/0000-0002-5939-752X
Brooke Puttergill https://orcid.org/0000-0002-1759-1033
REFERENCES
1. Neumann HPH, Young WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med 2019;381(6):552–565. DOI: 10.1056/nejmc1912022
2. Hu J, Wu J, Cai L, et al. Retroperitoneal composite pheochromocytoma-ganglioneuroma: a case report and review of literature. Diagn Pathol 2013;8:63. DOI: 10.1186/1746-1596-8-63
3. Thiel EL, Trost BA, Tower RL. A composite pheochromocytoma/ganglioneuroblastoma of the adrenal gland. Pediatr Blood Cancer 2010;54(7):1032–1034. DOI: 10.1002/pbc.22436
4. Lam AK. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr Pathol 2017;28(3):213–227. DOI: 10.1007/s12022-017-9484-5
5. Thompson LDR. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002;26(5):551–566. DOI: 10.1097/00000478-200205000-00002
6. Dhanasekar K, Visakan V, Tahir F, et al. Composite phaeochromocytomas—a systematic review of published literature. Langenbecks Arch Surg 2022;407(2):517–527. DOI: 10.1007/s00423-021-02129-5
7. Sandhu S, Jialal I. Glucagonoma syndrome. In: StatsPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021.
8. Contreras LN, Budd D, Yen TS, et al. Adrenal ganglioneuroma-pheochromocytoma secreting vasoactive intestinal polypeptide. West J Med 1991;154(3):334–337.
9. Onozawa M, Fukuhara T, Minoguchi M, et al. Hypokalemic rhabdomyolysis due to WDHA syndrome caused by VIP-producing composite pheochromocytoma: a case in neurofibromatosis type 1. Jpn J Clin Oncol 2005;35(9):559–563. DOI: 10.1093/jjco/hyi139
10. Negro A, Verzicco I, Tedeschi S, et al. Irreversible watery diarrhea, severe metabolic acidosis, hypokalemia and achloridria syndrome related to vasoactive intestinal peptide secreting malignant pheochromocytoma. Front Endocrinol (Lausanne) 2021;12:652045. DOI: 10.3389/fendo.2021.652045
11. Kikuchi Y, Wada R, Sakihara S, et al. Pheochromocytoma with histologic transformation to composite type, complicated by watery diarrhea, hypokalemia, and achlorhydria syndrome. Endocr Pract 2012;18(4):e91–e96. DOI: 10.4158/EP11370.CR
12. George DJ, Watermeyer GA, Levin D, et al. Composite adrenal phaeochromocytoma-ganglioneuroma causing watery diarrhoea, hypokalaemia and achlorhydria syndrome. Eur J Gastroenterol Hepatol 2010;22(5):632–634. DOI: 10.1097/MEG.0b013e328311a697
13. Shah MH, Goldner WS, Benson AB, et al. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2021;19(7):839–868. DOI: 10.6004/jnccn.2021.0032
14. Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2014;99(6):1915–1942. DOI: 10.1210/jc.2014-1498
15. Sbardella E, Grossman AB. Pheochromocytoma: an approach to diagnosis. Best Pract Res Clin Endocrinol Metab 2020;34(2):101346. DOI: 10.1016/j.beem.2019.101346
16. Garcia-Carbonero R, Matute Teresa F, Mercader-Cidoncha E, et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin Transl Oncol 2021;23(10):1995–2019. DOI: 10.1007/s12094-021-02622-9
17. Haissaguerre M, Courel M, Caron P, et al. Normotensive incidentally discovered pheochromocytomas display specific biochemical, cellular, and molecular characteristics. J Clin Endocrinol Metab 2013;98(11):4346–4354. DOI: 10.1210/jc.2013-1844
________________________
© The Author(s). 2022 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.